For teachers: The module reviews the study of the structure and function of the CFTR protein (Cystic Fibrosis Transmembrane Conductance Regulator). This membrane- anchored conductance regulation protein is subjected to genetic mutations that lead to the development of cystic fibrosis disease. Through this module we will learn both about bioinformatics tools and about the relation between the molecular levels (DNA, RNA and protein) and the macro level (tissue, organism) as well as the relationship between genotype and phenotype. The students will be exposed to new bioinformatics tools while using the fund of knowledge previously acquired throughout the course of biology classes.
The teachers might choose to begin the module with a brief review of the translation of biologic data from gene (DNA) to protein as described in the section “Biological background”. It is important to review the subject protein structure, going through the concepts of primary structure (the amino acids sequence), secondary structure (structural units such as sheets or helices), and the tertiary structure (the three dimensional folding that endows the protein with its structure), which endows the protein with its functionality. It is important to mention the role of structural domains and active sites, which are the platform for protein activity. A helpful resource can be found in Molecular Genetics and Biotechnology, by Orna Mokedi Shavit, p. 37-38, or any other textbook. Also, it is important to emphasize the relationship between changes in the DNA sequence, meaning the different mutation types, and their effects on the messenger RNA sequence and especially on the structure and function of the protein, and thus the phenotype at the cellular level and the organism.
Cystic fibrosis (CF) is one of the most prevalent disease causing genetic disorders, with an incidence of 1 in 2,500 births. It affects multiple organs in the body, including the lungs, pancreas, intestines and sex organs (Figure 1).
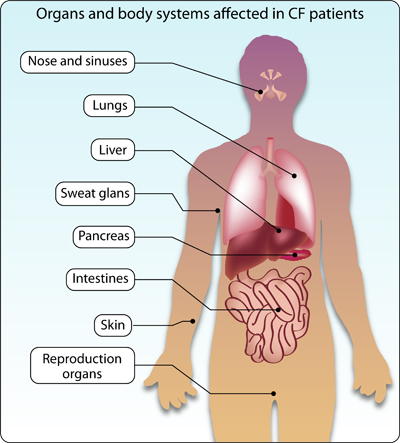 Figure 1: Characteristics of cystic fibrosis (CF)
Figure 1: Characteristics of cystic fibrosis (CF)
Cystic fibrosis is an autosomal recessive genetic disorder caused by a defect in the CFTR protein (Cystic Fibrosis Conductance Regulator). The protein is anchored to the cell membrane of epithelial cells where it forms a pump that facilitates the movement of ions through the membrane, especially chloride ions (Figure 2).
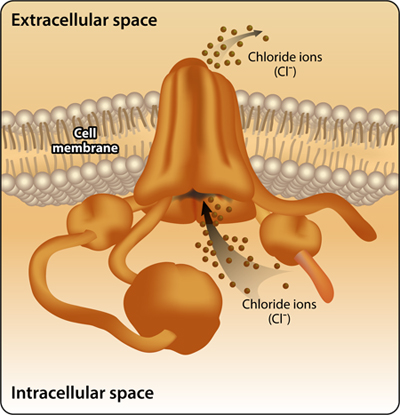 Figure 2: The CFTR protein functions as an ion channel in the cell membrane
Figure 2: The CFTR protein functions as an ion channel in the cell membrane
In healthy people, the epithelial cells secrete watery mucus that is aimed to protect the internal walls of the different organs from disease- causing agents. CF patients have an abnormal function of the ion- transporting channel, which results in a poor movement of chloride ions from inside the cell to the extracellular space. Due to an abnormally high osmotic pressure inside the cell, fluids are shifted into the cell and the mucus secreted from the epithelial cells is thicker and gluey. These thick secretions accumulate and obstruct the normal flow of fluids on one hand, and serve as a fertile ground for the proliferation of bacteria, and thus to the development of infections and inflammations (Figure 3).
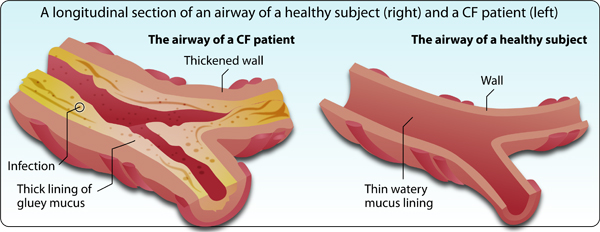
Figure 3: Thick mucus of the epithelial cells obstructs the airways and triggers infections in patients
Mutations in the CFTR gene interrupts the function of the ion channel in the cell membrane which leads to obstruction of tubal organs and development of infections and inflammations in various tissue and thus abnormal function of the different body systems (Figure 4).
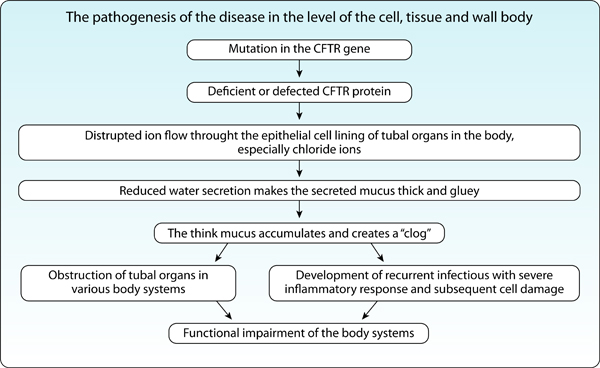 Figure 4: The sequence of events in CF, from a mutation in the CFTR gene to the functional impairment in body organs
Figure 4: The sequence of events in CF, from a mutation in the CFTR gene to the functional impairment in body organs
Increased awareness, early diagnosis, close surveillance and effective new therapies for CF have lead to longer life expectancy and improved patients’ quality of life. The significant progress in the understanding of the causes and pathogenesis of the disease, and the development of novel therapeutic modalities, promise CF patient a brighter future. Even though the disease was first diagnosed in 1938, the gene that encodes the CFTR protein was discovered only 50 years later; the Israeli researcher, Bat- Sheva Kerem , had a key role in its discovery. The discovery was a significant breakthrough in science and was even chosen as a milestone in genetics and genomics for the year 1989 by the prominent scientific journal, Nature (Figure 5).
 Figure 5: Discovering the gene for CFTR was chosen as a milestone in genetics for the year of 1989 by the prominent scientific journal Nature.
Figure 5: Discovering the gene for CFTR was chosen as a milestone in genetics for the year of 1989 by the prominent scientific journal Nature.
To date, over 1900 different disease- causing mutations were identified. The prevalence of the disease varies between different countries and ethnicities. In Israel, for example, CF is more prevalent among Ashkenazi Jews and is quite rare among Sephardic Jews. Also, in different areas, different mutations are prevalent. Mutations in the gene (DNA) lead to changes in the messenger RNA sequence and translate to changes in the protein sequence that affect its structure and function. The diagnosis of carriers of a mutant allele is based on the identification of DNA mutations. Choosing a treatment for a patient is based on the understanding of the relationship between the individual mutation and its effect on the protein structure and function.
As part of a national medical project aimed to understand the causes of cystic fibrosis and finding cure, researchers mapped and characterized CFTR gene mutations prevalent among the Israeli population. The researchers wish to learn how changes in the gene sequence affect protein structure and function, and then to be able to predict disease severity and choose individualized therapies according to the specific mutation a patient carries. Yet, before the researchers put on white coats and gloves and perform expensive lab experiments, plenty of valuable data can be acquired using bioinformatics tools.
In this module we will employ bioinformatics tools to study two main aspects of disease causing mutations: characterizing the protein defect and diagnosing carriers of mutated alleles. We will start by studying the structure of the CFTR protein and then see how different CFTR gene mutations alter its sequence and structure. We will also design experimental tools for diagnosing carriers of a mutant allele.
The steps to achieve the goals:
- First task: Studying protein structure – using the tool Prosite we will search for structural and functional motifs in CFTR.
- Second task: Diagnosis of carriers of a CFTR mutant allele – using the tool Primer3Plus we will design primers for PCR analysis.
For teachers: In the first task we will search for motifs of structural or functional importance in normal and mutant CFTR protein using the tool Prosite. The students will learn how to use the tool and interpret the results.
In the second task we will use the polymerase chain reaction (PCR) as a tool for the diagnosis and identification of allele mutations. We will design PCR primers for the different mutations using the tool Primer3Plus.
References to guided tutorials for these tools can be found in the module itself. These tutorials can also be taken before starting the module.
The students will study the effects of different CFTR gene mutations on the messenger RNA sequence, and on the sequence, structure and function of the protein. Thus, they will learn about the relation of genotype and phenotype. Finally, the students will design tools for the identification of the studied mutations. That way, they will pursue the pathway of understanding protein structure and function, continue to its relevance to disease phenotype, choice of therapies and drug development, and eventually, designing tools to screen for carriers.