Researchers have so far discovered over 1900 different mutations in the CFTR gene. In different countries and ethnic groups, different mutations are prevalent. The most common mutation worldwide, and in Israel as well, comprises almost two thirds of the mutant alleles, is called F508del. After having characterized the mutation at the DNA and RNA levels, researchers wish to examine how it affects the structure and function of the CFTR protein. The researchers ask: Can we predict, and to what extent, which motifs with structural or functional significance are affected by the mutation based on the sequence of the mutant protein? Can we predict disease severity based only on the sequence of the mutant protein? In order to answer these questions, the researchers use Prosite to analyze the common F508del mutation.
Teachers: At this point (or as part of the introduction), you can hold a class discussion about the different types of mutations (replacement, deletion, insertion etc.) and their position along the gene sequence (in exons, introns, CDS, UTR etc.) and how it might affect the protein sequence and structure.
In figure 4, presented are the DNA sequences of CFTR in a normal allele and the F508del mutant allele, and the proteins they encode.
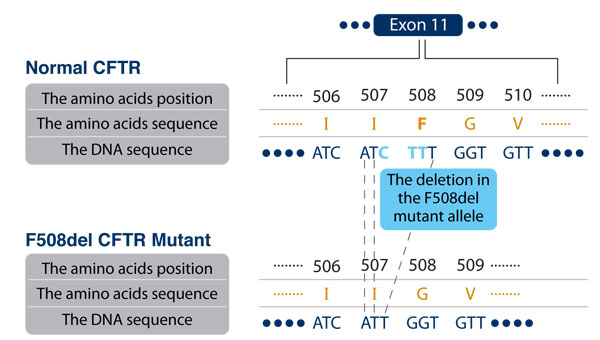
Figure 4: the sequences of nucleotides (in blue) and the amino acids (in brown) in a normal and F508del mutated CFTR. The nucleotides that are missing in the mutant allele are marked (in light blue).
Teachers: There is another common mutation in which the missing nucleotides are TCT, which means that the deletion starts one position earlier. The resulting DNA and protein sequences are similar to those resulting from CTT deletion and thus this mutant is also designated F508del.
8. Analyzing to the data presented in figure 4, what can we learn about the effect of the F508del mutation on the protein?
- A 3 nucleotides deletion in exon 11 leads to a replacement of the amino acid phenylalanine (symbol – F) in position 508 with the amino acid glycine (G) in the protein.
- A 3 nucleotides deletion in exon 11 forms a protein sequence that misses the amino acid phenylalanine (F) in position 508.
- The 3 nucleotides deleted in exon 11 encompass two codons; therefore, the mutant CFTR protein will harbor a change of two amino acids, coded by these two codons.
- The deletion mutation in exon 11 leads to a frame shift change, which will completely change the protein sequence from position 508.
The answer is: B. The mutant allele misses 3 nucleotides in exon 11 (CTT, Figure 4). The codons ATC CTT in the normal allele encode two amino acids, I in position 507 and F in position 508 (I507, F508), but are changed in the mutant allele to one codon , ATT, that encodes only the amino acid, I507. This explains the name F508del for the mutation: the amino acid phenylalanine (symbol – F) in position 508 is missed (“del” – a short for deletion).
9. Does the deletion mutation, F508del, change the reading frame?
- The reading frame does not change – 3 nucleotides are missing and the genetic code is read in triplets (codons), so translation can continue normally from codon GTT and on.
- The reading frame changes because the missing nucleotides encompass two different codons.
- The reading frame does not change because in position 507, both in the normal and mutant alleles, the amino acid isoleucine (I) appears.
- The reading frame changes – only a mutation in which one nucleotide is replaced with another does not change the reading frame. Insertions and deletions change the reading frame.
The answer is: A. The reading frame does not change because three nucleotides are missing and the protein sequence will be the same starting from position 509 in the CFTR protein.
10. Why is the amino acid I in position 507 in the mutant protein not affected despite the change in its coding codon?
- Because only the left and central positions in a codon code for the amino acid and they did not change.
- Since the reading frame did not change, there will not be changes in amino acids at all.
- Both codons, ATC in the normal allele and the mutated ATT, encode the same amino acid, isoleucine, which symbol is I.
- Since three nucleotides are missing, the length of a single codon, only one position is affected – position 508 and not 507.
The answer is: C. Both codons, ATC in the normal allele and the mutated ATT, encode the same amino acid, isoleucine (I).
In order to determine which protein motifs are affected by the mutation, we can analyze it using Prosite, and compare the search results obtained for the F508del mutant protein with those of the normal CFTR protein. This method is called comparative analysis. Here is the amino acid sequence of F508del CFTR.
Lets open a new web browser tab (Ctrl+N) and enter Prosite again, without closing the search results tab we have for the normal CFTR. Reminder: http://www.expasy.org/prosite). Copy the F508del CFTR sequence, paste in the designated window and click “Scan” in order to begin searching. Now lets compare the results obtained for the F508del mutant protein with those obtained for the normal protein. Pay attention to the score that measures the sequence similarity between the sequences as appear in the query protein and the motif as defined in the database.
11. Review the results page for the F508del mutant protein. Did Prosite identify 4 motifs in this protein sequence?
- Yes
- No
The answer is: A. Yes.
At a first glance, it seems that the mutation did not affect protein structure, and that the results that were obtained for the normal protein and the F508del mutant protein are identical. Lets look carefully at the two results pages and compare the scores for each motif in the normal protein with their counterparts in the mutant protein.
12. Which of the motifs in the F508del mutant protein is affected by the mutation and how?
- The first ATP binding motif ABC_TRANSPORTER_2 (at the N terminal end, beginning of the protein). Its score is higher in the mutant protein than in the normal protein.
- The second ATP binding motif ABC_TRANSPORTER_2 (at the C terminal end, end of the protein). Its score is higher in the mutant protein than in the normal protein.
- The first ATP binding motif ABC_TRANSPORTER_2 (at the N terminal end, beginning of the protein). Its score is lower in the mutant protein than in the normal protein.
- The second ATP binding motif ABC_TRANSPORTER_2 (at the C terminal end, end of the protein). Its score is lower in the mutant protein than in the normal protein.
The answer is: C. The first ATP binding motif is the one affected by the F508del mutation. The motif consists of amino acids in positions 423-646, including the one in position 508. Since the mutation affects the amino acid phenylalanine (F) in position 508, this motif is expected to be affected.
13. What does the change in the score for the motif in the mutant protein (compared with the score for the motif in the normal protein) mean?
- Decreased score means that the sequence in the mutant protein is less similar to the motif sequence as appears in the database.
- Decreased score means that the sequence in the mutant protein is more similar to the motif sequence as appears in the database.
- Increased score means that the sequence in the mutant protein is less similar to the motif sequence as appears in the database.
- Increased score means that the sequence in the mutant protein is more similar to the motif sequence as appears in the database.
The answer is: A. The higher the score is, the higher is the sequence similarity between the sequence in the query protein and the sequence of the motif as is deposited in the database. The mutation F508del leads to a decrease in the score calculated for the first ATP binding motif, thus we can assume that due to the mutation the sequence of the motif in the mutant protein is less similar to the sequence of the motif in the database.
Teachers: Answers A and D are both basically correct but in our case the mutation leads to a decreased similarity score, and not increased, and thus answer A is the best choice.
A basic assumption in bioinformatics is that homology between amino acid sequences may reflect three-dimensional structural and functional similarities between proteins. Sequences that are essential for the structure or function of proteins are usually evolutionary conserved and accumulated fewer mutations throughout evolution. In fact, in order to determine a sequence for a particular motif, Prosite compares conserved segments of protein sequences which structure or function is already established. When scanning a query sequence through the motif database, higher scores are given if higher sequence similarities are obtained.
Even though the decrease in score is not so significant, given that only one amino acid out of over 220 amino acids in the motif is missing, it is often enough to impair the protein function. In this case, it was found that due to the missing amino acid, phenylalanine in position 508, the protein does not fold properly, which leads to its degradation. As a result, there is almost no active protein available at the cell membrane.
We can now map the effect of the F508del mutation on the protein motifs in CFTR (Figure 5).
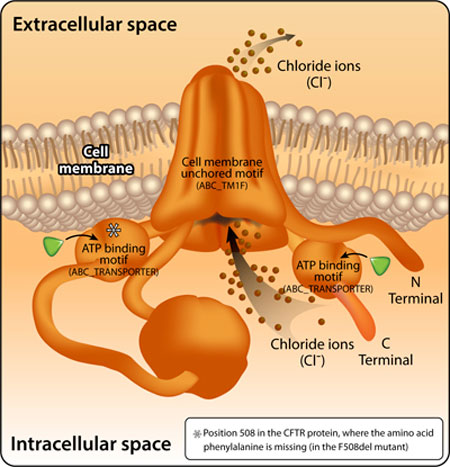
Figure 5: The position of the missing amino acid in the F508del mutant CFTR protein.
Prosite – based analysis revealed that the amino acid phenylalanine in position 508 is part of the ATP binding motif (ABC_TRANSPORTER_2) at the N terminal end of the protein (the beginning of the sequence). Due to a mutation that causes an amino acid deficiency in this position, compared to the motif in the normal protein, the motif sequence in the mutant protein is less similar to the sequence that appears in the database, reflected by a lower similarity score. The change in sequence leads to an abnormal folding of the protein and its degradation. Because of very low production of the normal protein that is active in the cell membrane, the patients suffer from severe symptoms of the disease.